챕터 03 선수지식: 차세대 시퀀싱에 들어가기 전 알아야 할 것
1. 3챕터는 무엇을 하려는 장인가요?
3챕터는 차세대 시퀀싱, 즉 NGS의 원리와 데이터 구조를 설명하는 장입니다. 여기서는 Illumina, Nanopore, PacBio, Short Read, Long Read, 라이브러리, 어댑터, 플로우 셀, Read 1/Read 2, FASTQ, Phred 품질 점수, 정렬 알고리즘, BLAST, k-mer, BWT, FM Index, SAM, CIGAR 같은 개념이 등장합니다.
2챕터 선수지식에서 DNA를 A/T/G/C 문자열로 보는 관점을 이미 다뤘습니다. 3챕터에서는 그 DNA 문자열을 실제 장비가 어떻게 읽고, 컴퓨터가 그 조각들을 어떻게 처리하는지 이해해야 합니다.
3챕터를 관통하는 핵심 질문은 이것입니다.
긴 DNA를 어떻게 읽어서 컴퓨터가 분석 가능한 파일로 만들까요?
2. 시퀀싱은 DNA 글자를 읽는 기술입니다
시퀀싱(Sequencing)은 DNA나 RNA의 염기서열을 읽는 기술입니다. 즉, A, T, G, C가 어떤 순서로 배열되어 있는지 알아내는 과정입니다.
DNA를 매우 긴 책이라고 생각해보겠습니다. 이 책을 처음부터 끝까지 한 번에 읽을 수 있으면 좋겠지만, 실제 기술은 보통 그렇게 단순하지 않습니다. 특히 NGS는 긴 DNA를 잘게 자른 다음, 많은 조각을 동시에 읽고, 그 결과를 컴퓨터로 다시 해석합니다.

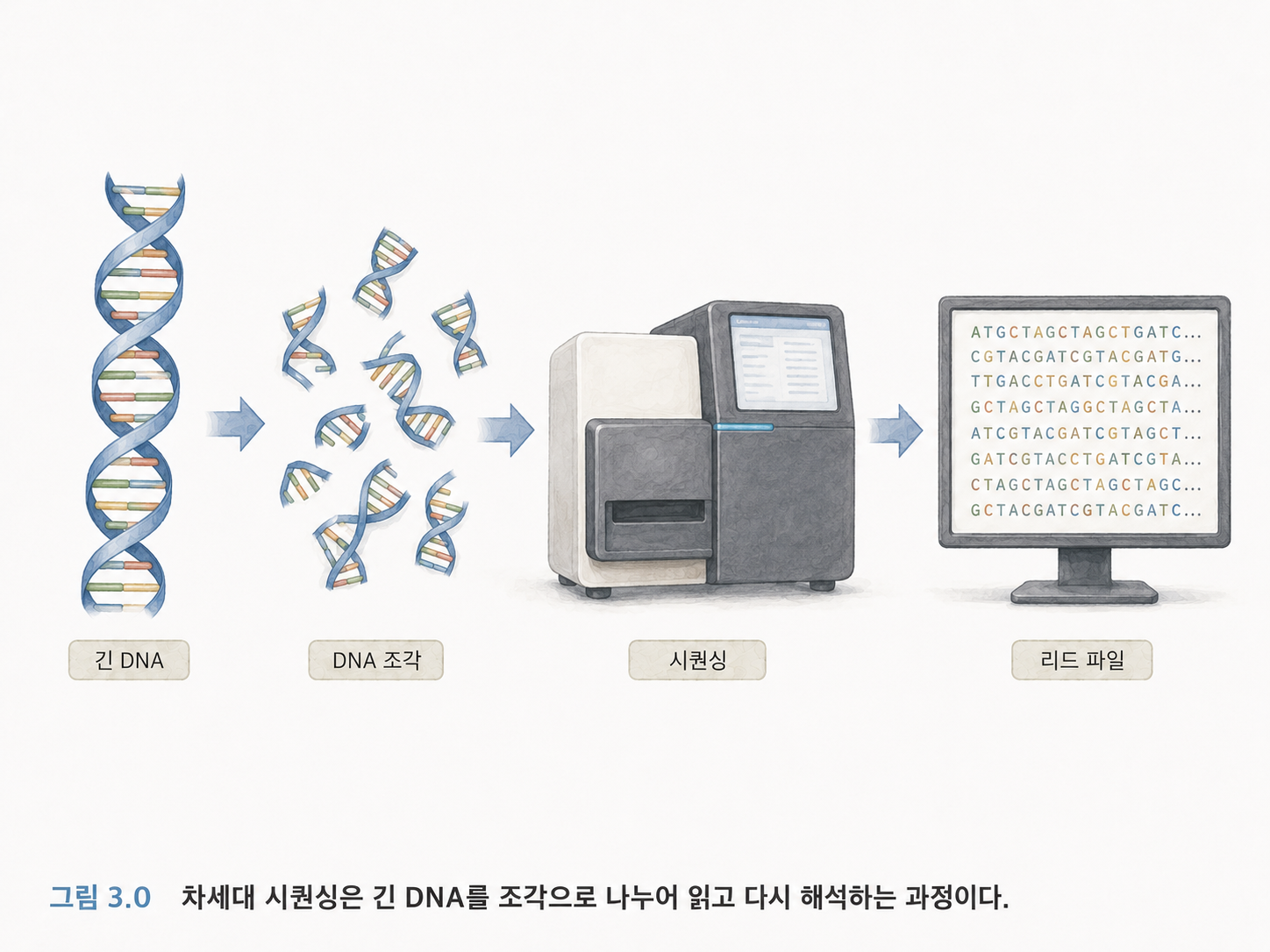
여기서 “리드(Read)”라는 말이 중요합니다. 리드는 시퀀싱 장비가 읽어낸 DNA 조각의 서열입니다. 원본 DNA 전체가 아니라, 그중 일부 조각을 읽은 결과입니다.
예를 들어 원래 DNA가 아주 긴 문장이라면, 리드는 그 문장에서 잘라낸 짧은 구절입니다.
원본 DNA: ATGCTAGGCTTACGATCGATCGT...
리드 1: ATGCTAGG
리드 2: GGCTTACG
리드 3: ACGATCGA
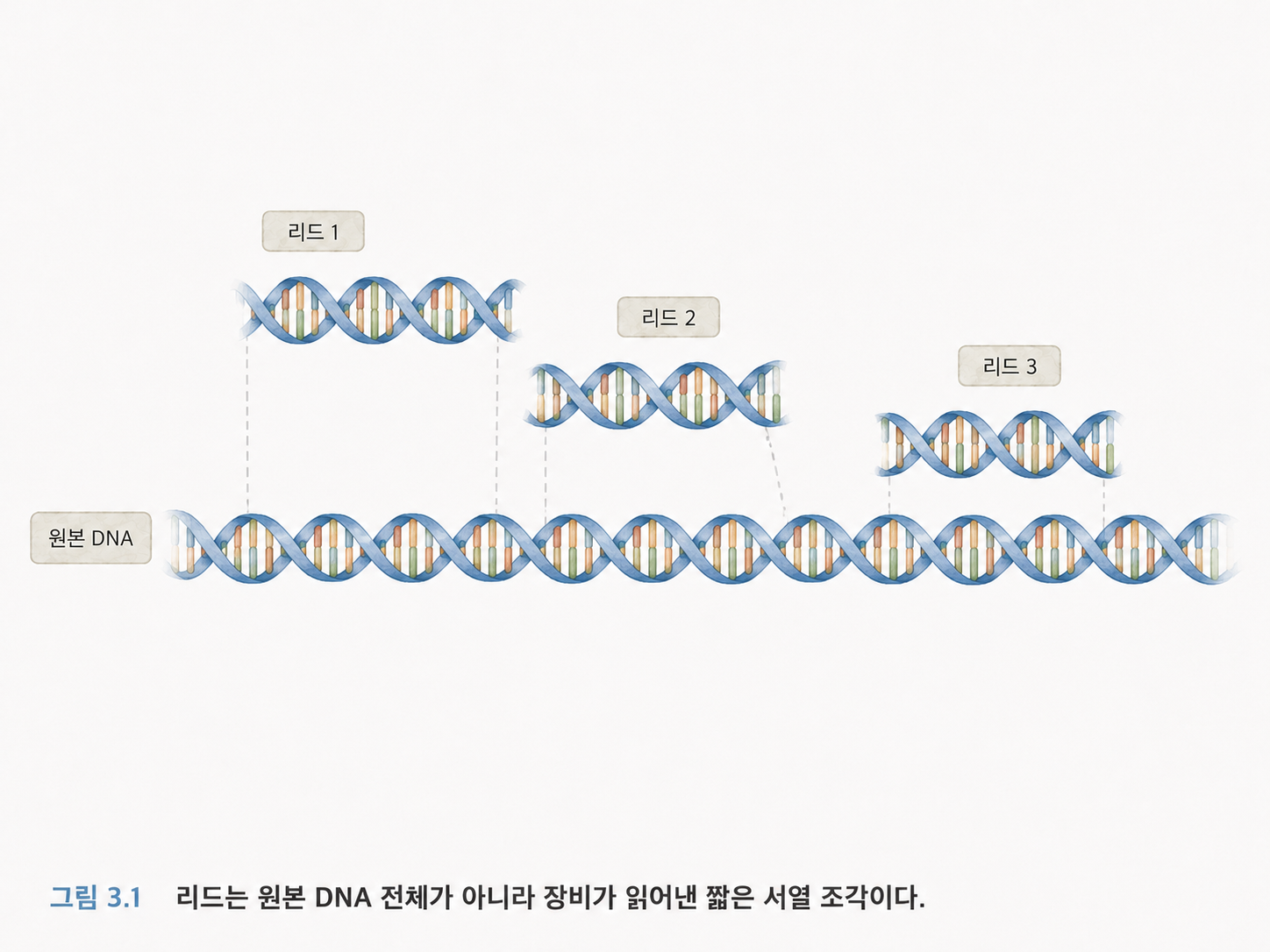
컴퓨터는 이런 리드들을 모아서 원래 위치를 추정하거나, 표준 유전체에 붙여서 변이를 찾습니다.
3. Short Read와 Long Read는 읽는 조각의 길이가 다릅니다
NGS 기술을 이해할 때 Short Read와 Long Read의 차이를 알아야 합니다.
Short Read는 짧은 DNA 조각을 매우 정확하게 많이 읽는 방식입니다. Illumina가 대표적입니다. 짧은 문장을 많이 읽는 방식이라고 볼 수 있습니다.
Long Read는 훨씬 긴 DNA 조각을 읽는 방식입니다. Nanopore와 PacBio가 대표적입니다. 긴 문장을 읽을 수 있으므로 반복되는 구간이나 큰 구조 변이를 이해하는 데 유리합니다.
비유하면 이렇습니다.
| 구분 | 비유 | 장점 | 약점 |
|---|---|---|---|
| Short Read | 책을 짧은 문장 조각으로 많이 읽기 | 정확도가 높고 많이 생산하기 좋습니다. | 반복 구간이나 큰 구조 파악이 어렵습니다. |
| Long Read | 긴 문단 단위로 읽기 | 구조를 파악하기 쉽습니다. | 비용, 처리량, 오류 특성에서 기술별 차이가 있습니다. |
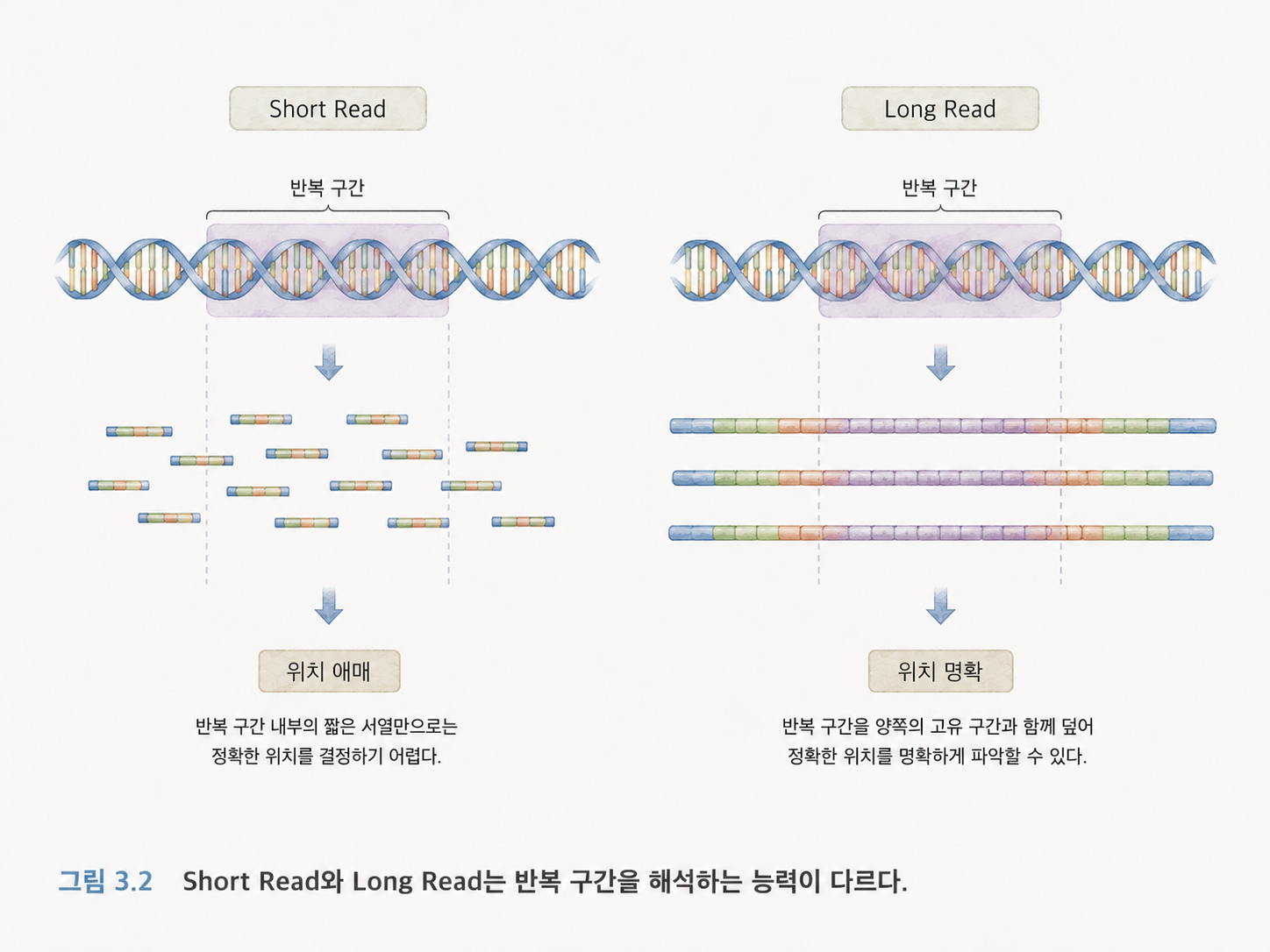
실제 분석에서는 Short Read와 Long Read를 함께 쓰기도 합니다. 짧은 리드는 정확도를 보완하고, 긴 리드는 구조를 파악하는 데 도움을 줍니다.
4. PCR은 DNA를 복사해서 신호를 키우는 과정입니다
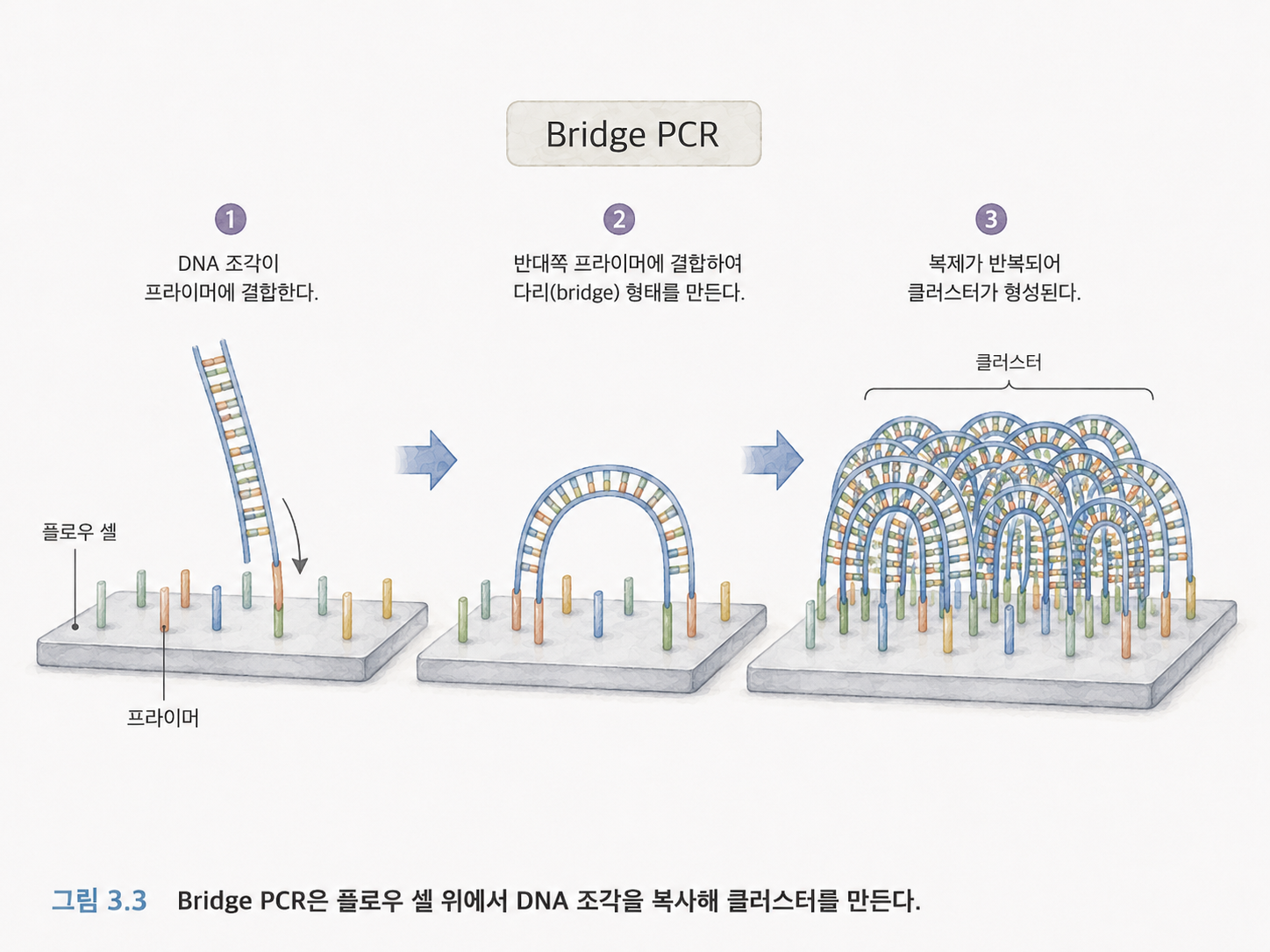
3챕터의 Illumina 설명에서 Bridge PCR이 등장합니다. PCR은 DNA를 많이 복사하는 기술입니다. 원본 DNA 조각이 하나만 있으면 장비가 신호를 읽기 어렵습니다. 그래서 같은 조각을 많이 복사해서 신호를 키웁니다.
Bridge PCR은 Illumina 플로우 셀 표면에서 DNA 조각이 다리처럼 휘어 붙고, 복사되고, 다시 분리되는 과정이 반복되어 클러스터를 만드는 방식입니다. 클러스터는 같은 DNA 조각의 복사본들이 모여 있는 작은 무리입니다.
처음에는 세부 화학 과정을 모두 외울 필요는 없습니다. 핵심은 이것입니다.
Illumina는 DNA 조각을 플로우 셀 위에 붙이고, 같은 조각을 많이 복사해서, 형광 신호를 읽기 충분할 만큼 증폭합니다.
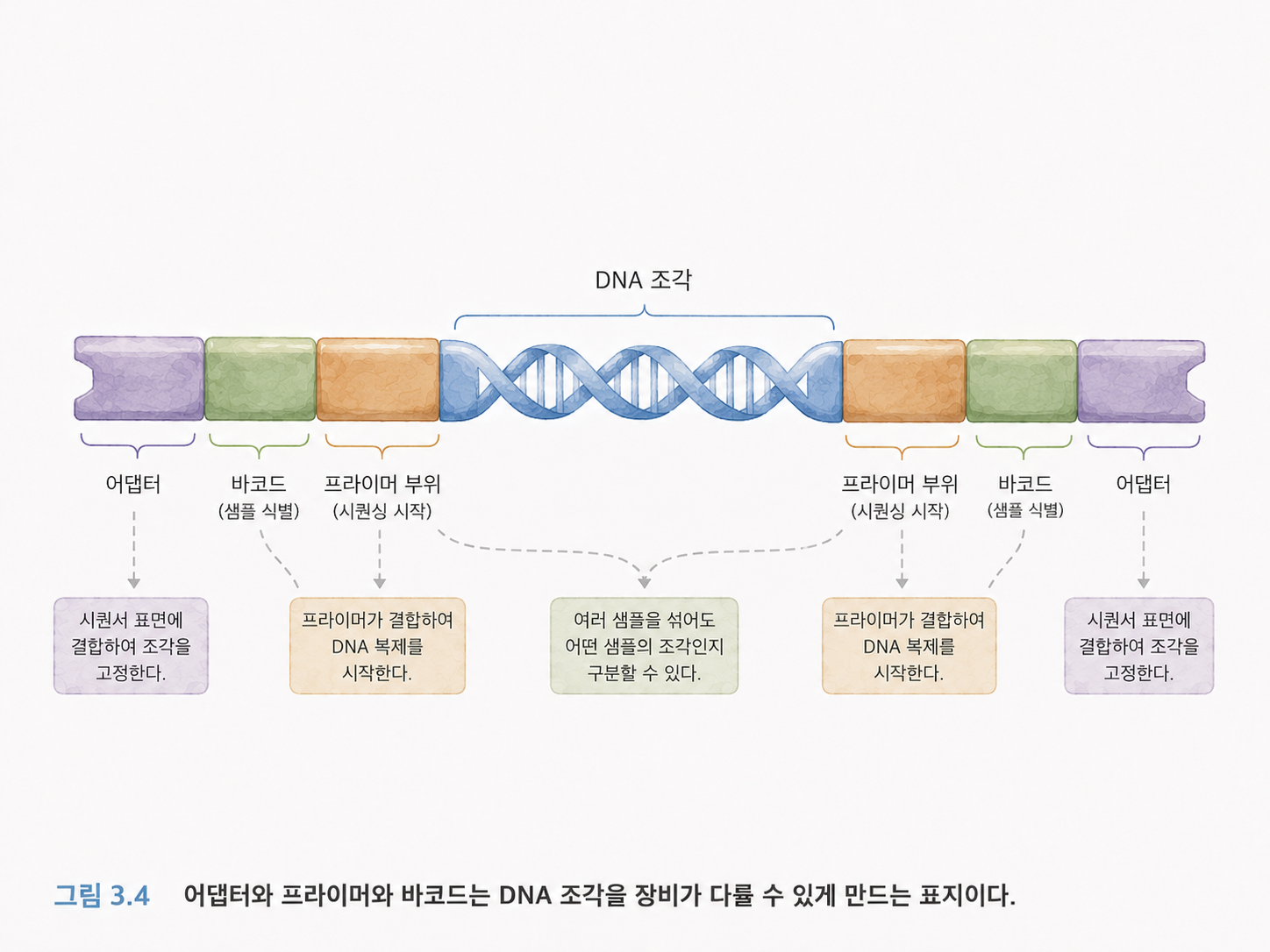
5. 어댑터와 프라이머는 DNA 조각에 붙이는 손잡이입니다
NGS에서는 DNA 조각을 그냥 장비에 넣지 않습니다. 먼저 시퀀싱 라이브러리라는 형태로 준비합니다. 이때 DNA 조각 양끝에 어댑터라는 짧은 서열을 붙입니다.
어댑터는 손잡이처럼 생각하면 됩니다. 장비가 DNA 조각을 붙잡고, 복사하고, 읽기 시작하려면 정해진 부착 부위가 필요합니다. 그 역할을 어댑터가 합니다.
프라이머는 DNA 합성이 시작되는 출발점입니다. 복사를 시작하려면 “여기서부터 읽으세요”라고 알려주는 짧은 서열이 필요한데, 그것이 프라이머입니다.
인덱스 또는 바코드는 여러 샘플을 섞어서 한 번에 시퀀싱한 뒤, 나중에 어느 샘플에서 온 리드인지 구분하게 해주는 꼬리표입니다. 택배 상자에 붙은 송장 번호처럼 생각하면 됩니다.
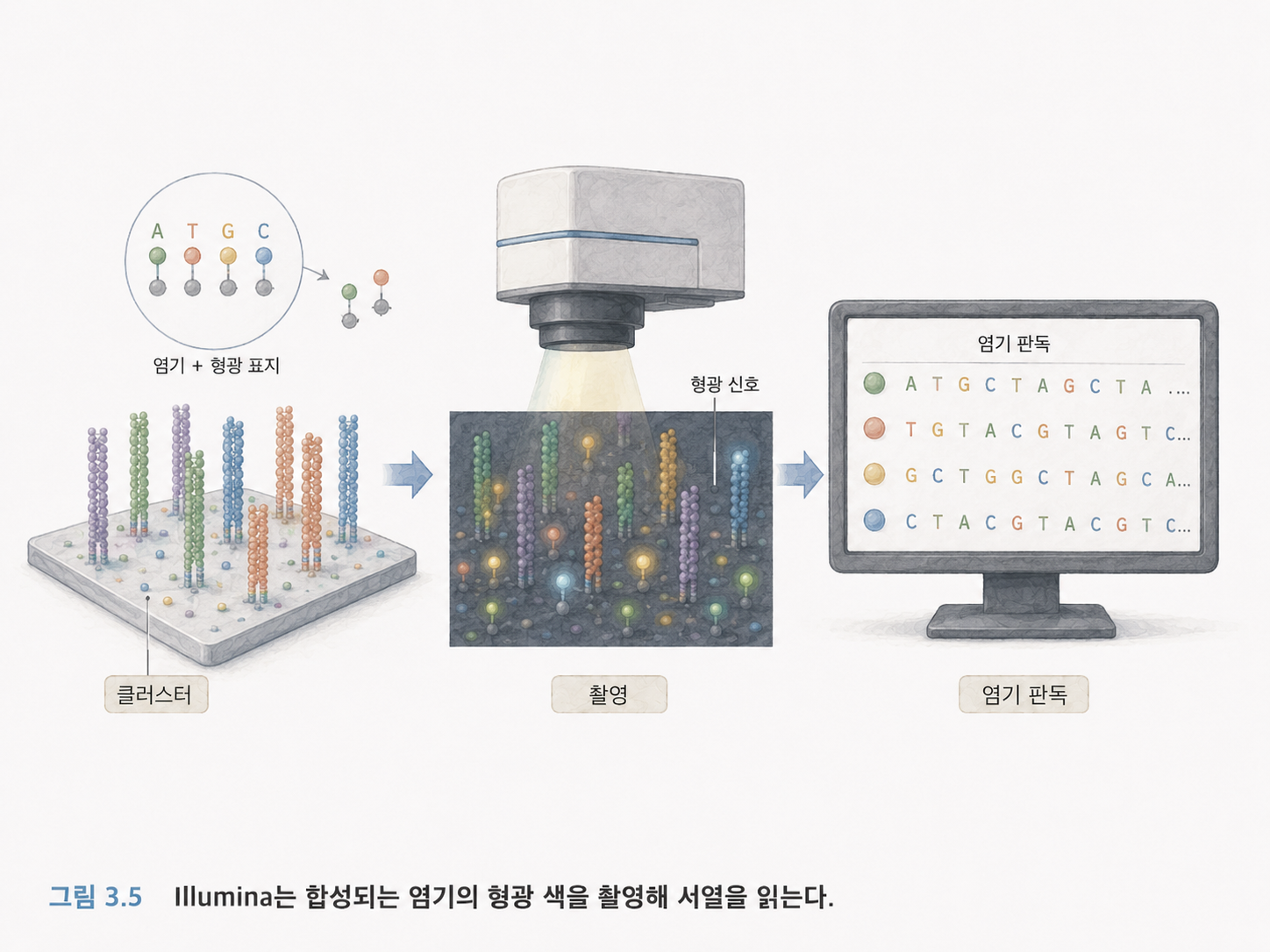
6. Illumina는 형광 신호로 염기를 읽습니다
Illumina의 핵심 원리는 합성에 의한 시퀀싱입니다. DNA를 한 글자씩 합성하면서, 새로 들어온 염기가 A인지 T인지 G인지 C인지 형광 색으로 확인합니다.
아주 단순화하면 다음 흐름입니다.
- DNA 조각을 플로우 셀에 붙입니다.
- Bridge PCR로 같은 조각의 복사본 무리를 만듭니다.
- 형광 표지가 붙은 염기를 하나씩 넣습니다.
- 어떤 색이 빛났는지 사진을 찍습니다.
- 그 색을 보고 A/T/G/C 중 무엇이 들어왔는지 판단합니다.
- 이 과정을 반복해 리드를 만듭니다.
즉, Illumina 시퀀싱은 “화학 반응 + 형광 촬영 + 컴퓨터 판독”의 조합입니다.
7. Nanopore는 전류 변화를 읽습니다
Nanopore 시퀀싱은 Illumina와 방식이 많이 다릅니다. DNA가 아주 작은 구멍, 즉 나노포어를 통과할 때 전류가 변합니다. 염기 종류에 따라 전류 변화 패턴이 달라지므로, 그 신호를 해석해서 염기서열을 추정합니다.
쉽게 말하면, 좁은 문을 지나가는 사람의 모양에 따라 문에 생기는 흔들림이 달라지는 것과 비슷합니다. Nanopore는 DNA 글자가 구멍을 지나갈 때 생기는 전기 신호의 차이를 읽습니다.
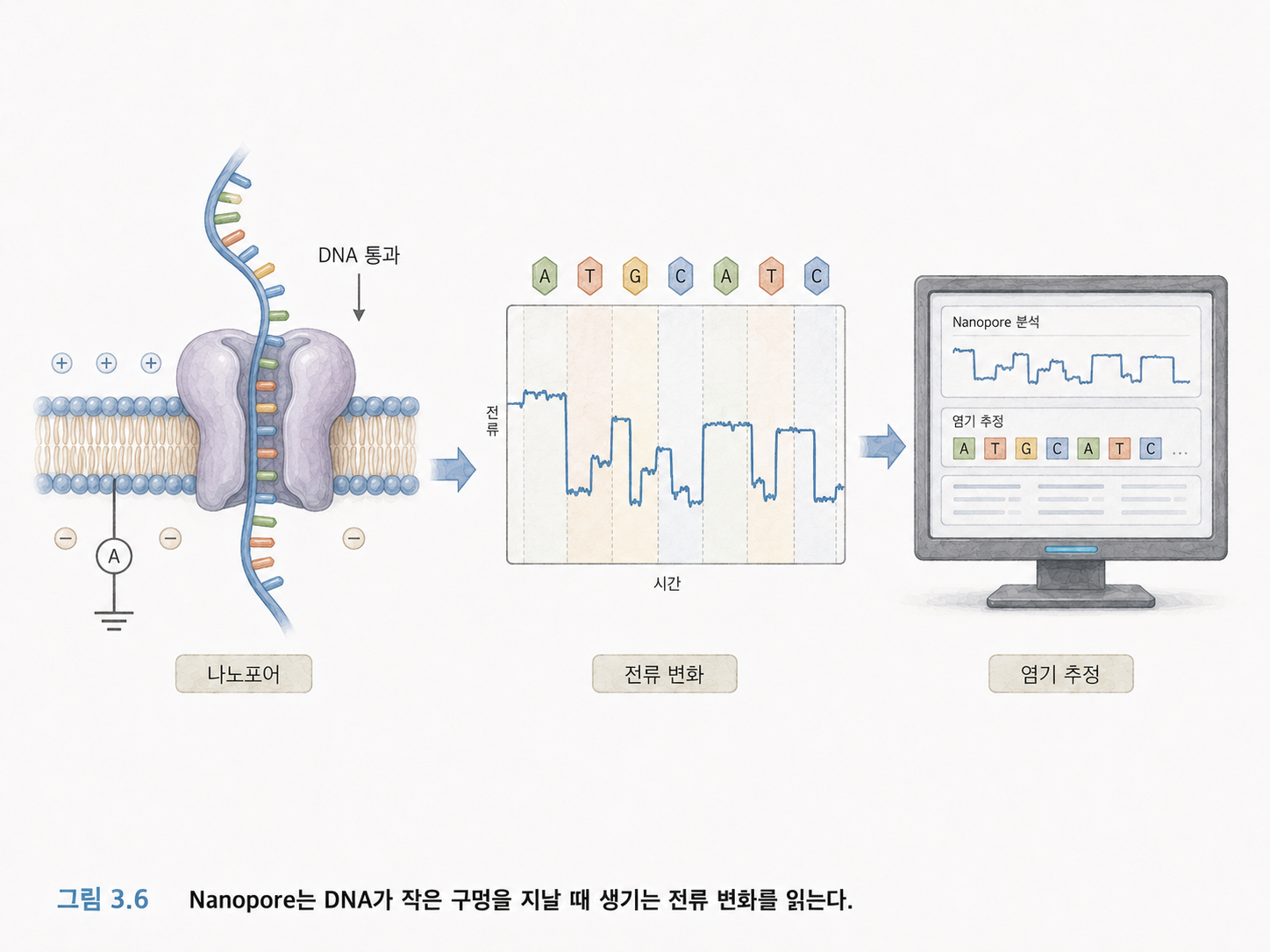
Nanopore의 장점은 긴 리드를 읽을 수 있다는 점입니다. 매우 긴 DNA 조각을 읽을 수 있어 구조적 변이나 반복 서열 분석에 유리합니다.
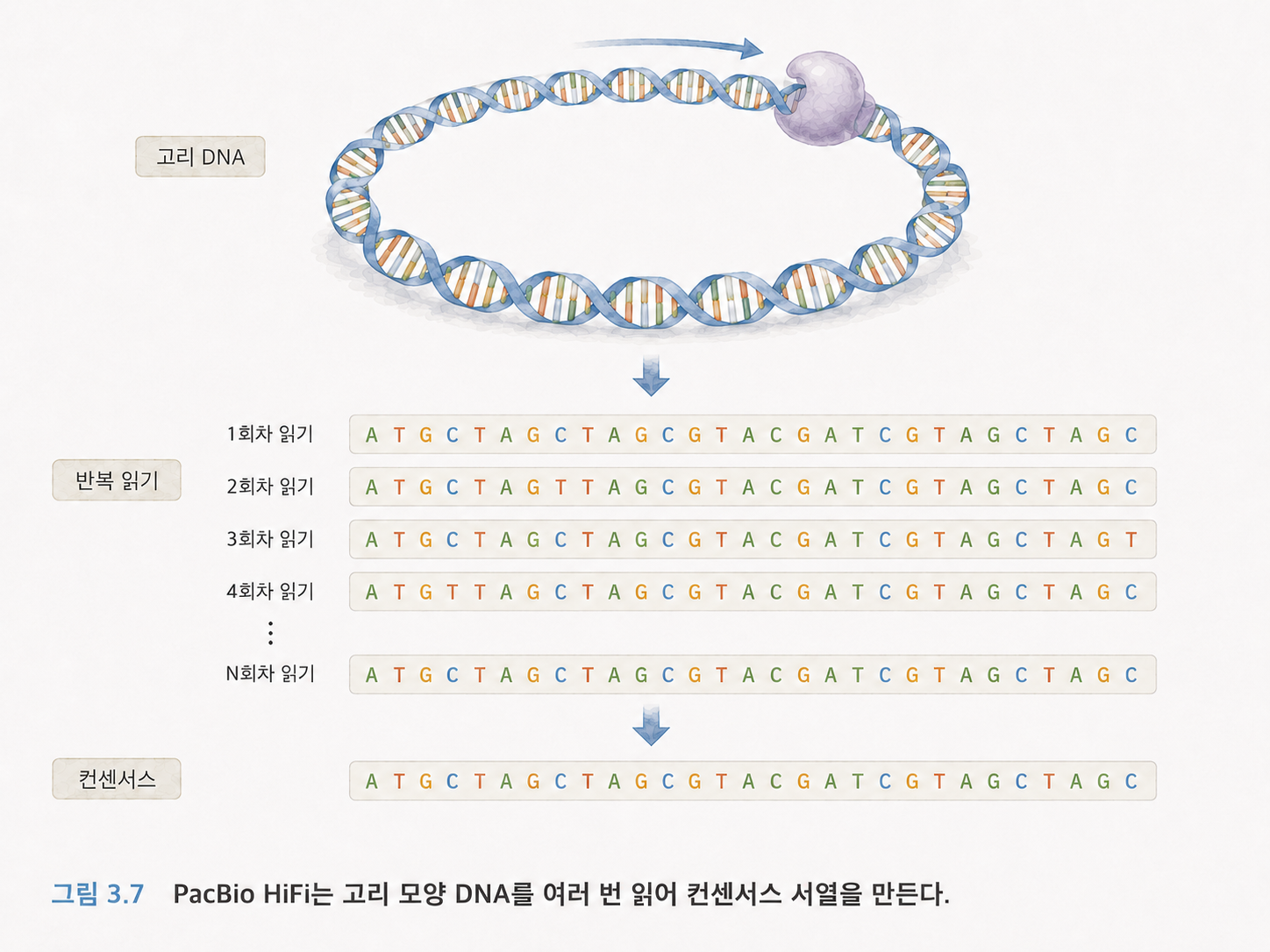
8. PacBio는 같은 긴 조각을 여러 번 읽어 정확도를 높입니다
PacBio는 단일 분자 실시간 시퀀싱 기술을 사용합니다. 특히 HiFi 시퀀싱은 DNA 조각을 고리 모양으로 만들어 같은 구간을 여러 번 읽고, 그 결과를 합쳐 정확한 컨센서스 서열을 만듭니다.
컨센서스는 여러 번 읽은 결과를 종합해 가장 그럴듯한 최종 답을 만드는 것입니다. 여러 사람이 같은 문장을 받아쓰기했을 때, 다수의 결과를 비교해 오타를 줄이는 과정과 비슷합니다.
PacBio HiFi는 Long Read의 장점과 높은 정확도를 함께 노리는 기술이라고 이해하면 됩니다.
9. 유전체 조립은 퍼즐 맞추기입니다
유전체 조립(Genome Assembly)은 잘게 읽은 DNA 조각들을 이용해 원래의 긴 유전체 서열을 재구성하는 작업입니다.
퍼즐을 생각하면 쉽습니다. 원래 그림은 하나인데, 조각이 많이 흩어져 있습니다. 각 조각의 겹치는 부분을 찾아 원래 그림을 복원해야 합니다.
DNA 리드도 마찬가지입니다. 리드끼리 겹치는 서열을 이용해 원래 DNA를 추정할 수 있습니다. 다만 실제 유전체에는 반복되는 구간이 많아서 어렵습니다. 예를 들어 책 속에 “아주 아주 아주” 같은 반복 표현이 계속 나오면, 짧은 조각만 보고 어느 위치인지 구분하기 어렵습니다.
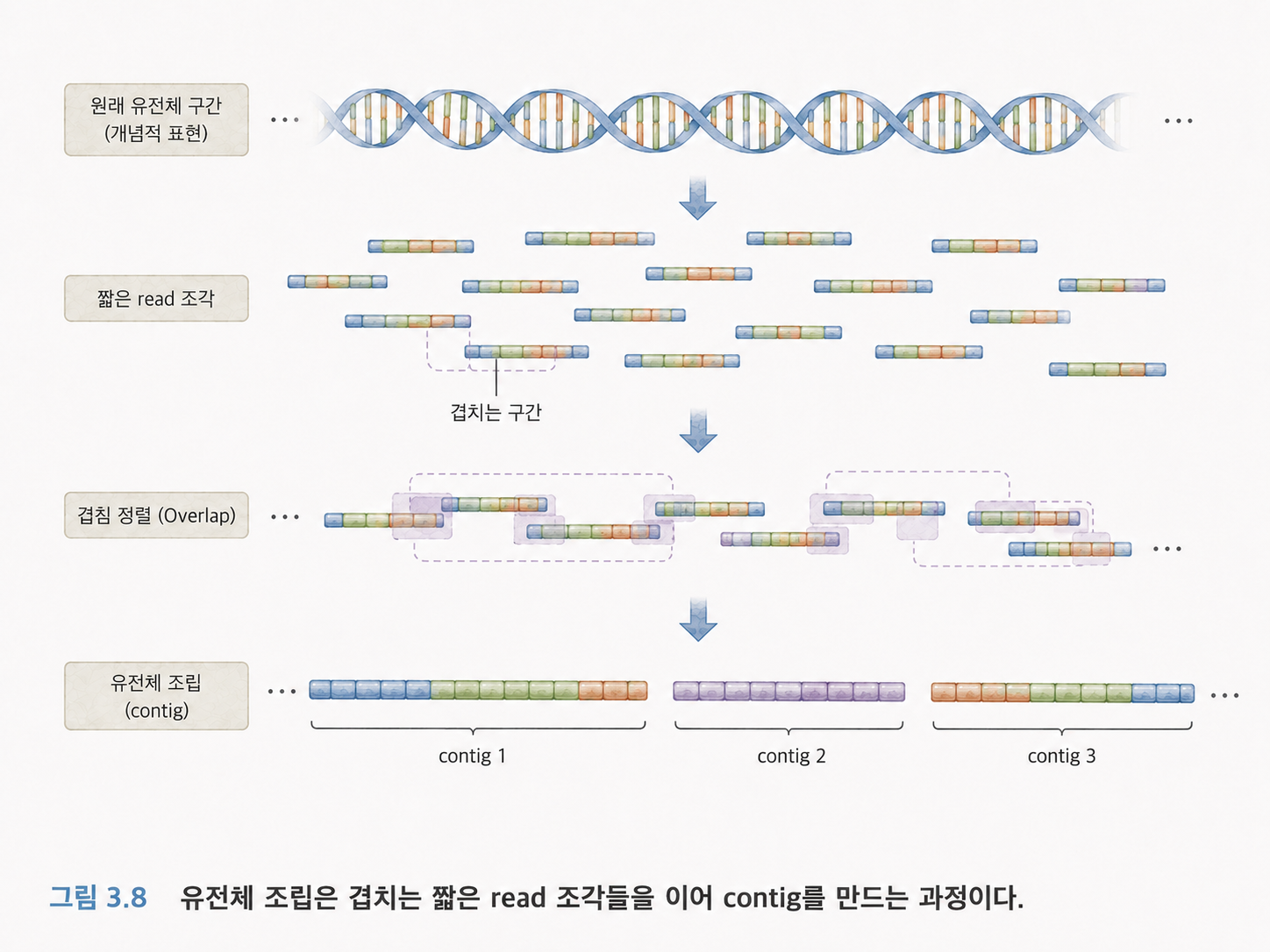
그래서 Long Read가 유전체 조립에 도움이 됩니다. 긴 조각은 반복 구간을 가로질러 읽을 수 있기 때문입니다.
10. FASTQ는 리드와 품질을 함께 담는 텍스트 파일입니다
3챕터에서 FASTQ 파일이 등장합니다. FASTQ는 시퀀싱 결과를 저장하는 대표적인 파일 형식입니다.

FASTQ에서 리드 하나는 보통 네 줄로 표현됩니다.
@read_id
ATGCAAGTCC...
+
IIIIHHHFGG...
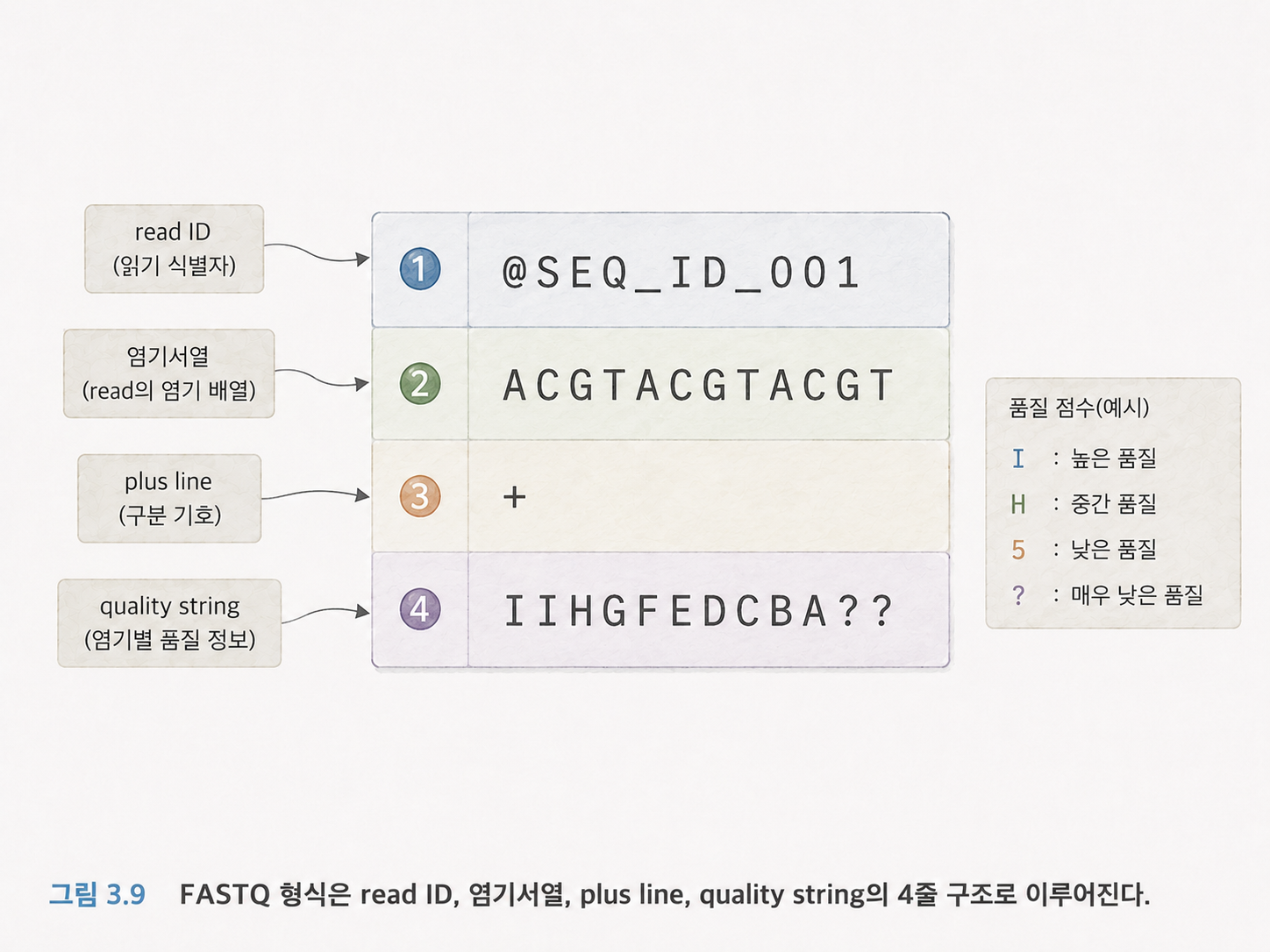
첫 번째 줄은 리드의 이름 또는 식별자입니다. 두 번째 줄은 실제로 읽은 DNA 서열입니다. 세 번째 줄은 구분선입니다. 네 번째 줄은 각 염기의 품질 점수입니다.
여기서 품질 점수는 “이 염기를 제대로 읽었을 가능성이 얼마나 높은가”를 나타냅니다. 시퀀싱 장비도 완벽하지 않기 때문에, 각 글자마다 신뢰도를 함께 기록합니다.
11. Phred 점수는 오류 확률을 표현하는 방식입니다
FASTQ의 품질 점수는 Phred 점수라는 방식으로 표현됩니다. 수식은 원문에 나오지만, 처음부터 수식을 외울 필요는 없습니다.
핵심은 이렇습니다.
- Q10은 오류 확률이 약 10%입니다.
- Q20은 오류 확률이 약 1%입니다.
- Q30은 오류 확률이 약 0.1%입니다.
- Q40은 오류 확률이 약 0.01%입니다.
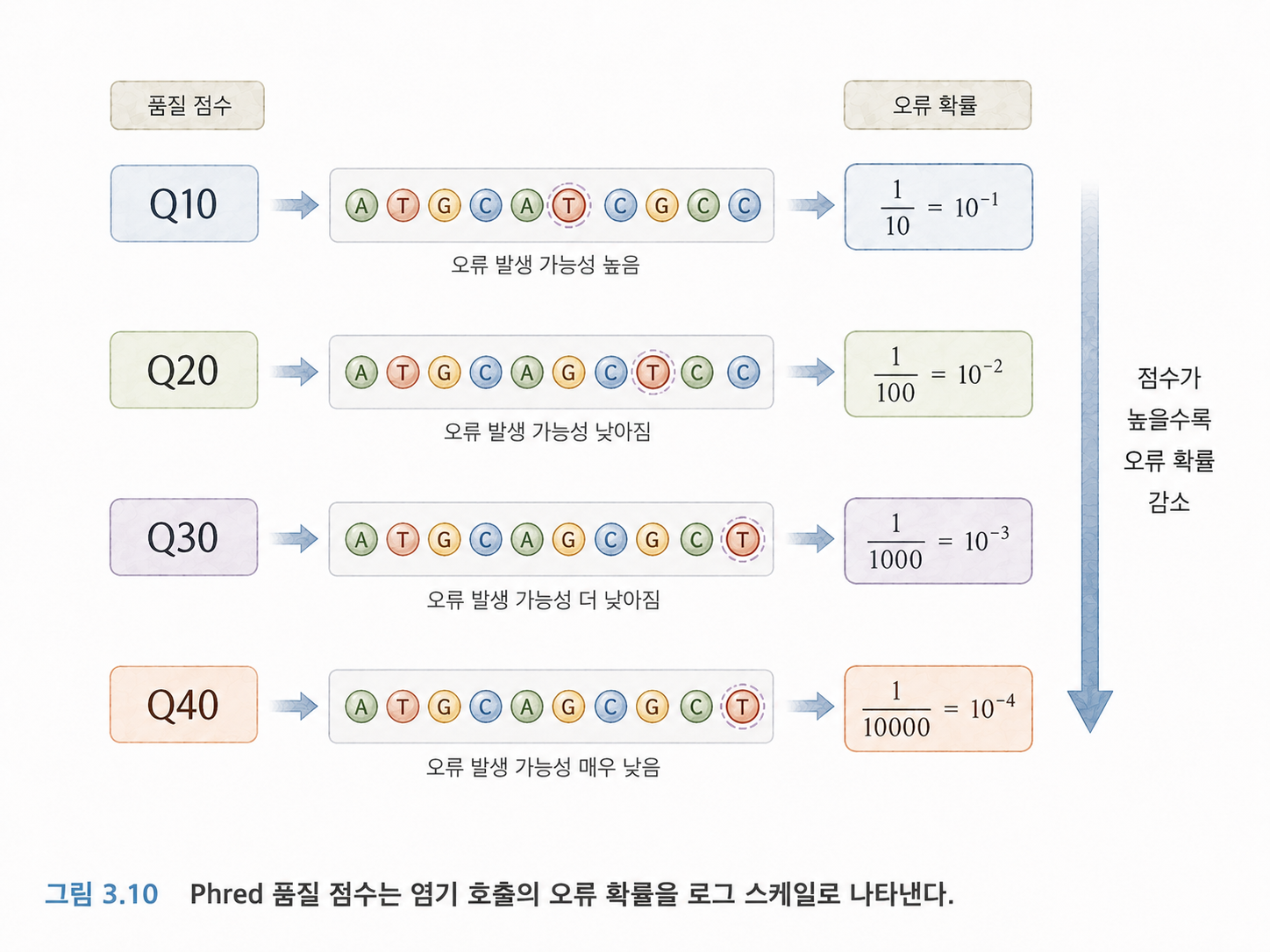
즉, 숫자가 높을수록 더 믿을 만한 염기 호출입니다. Q30은 생명정보학에서 흔히 고품질 기준처럼 자주 언급됩니다.
FASTQ 파일에서는 이 점수를 숫자 그대로 쓰지 않고 ASCII 문자로 인코딩합니다. ASCII는 컴퓨터에서 문자를 숫자로 표현하는 약속입니다. 3챕터에서 이 부분이 나오면, “품질 점수를 문자로 압축해서 적어둔 것”이라고 이해하면 됩니다.
12. 싱글 엔드와 페어드 엔드는 읽는 방향의 차이입니다
싱글 엔드 시퀀싱은 DNA 조각의 한쪽 끝만 읽는 방식입니다. 페어드 엔드 시퀀싱은 같은 DNA 조각의 양쪽 끝을 읽는 방식입니다.
페어드 엔드에서는 보통 R1 파일과 R2 파일이 생깁니다. R1은 한쪽 끝에서 읽은 리드들이고, R2는 반대쪽 끝에서 읽은 리드들입니다. 두 파일의 n번째 리드는 같은 DNA 조각에서 나온 짝입니다.
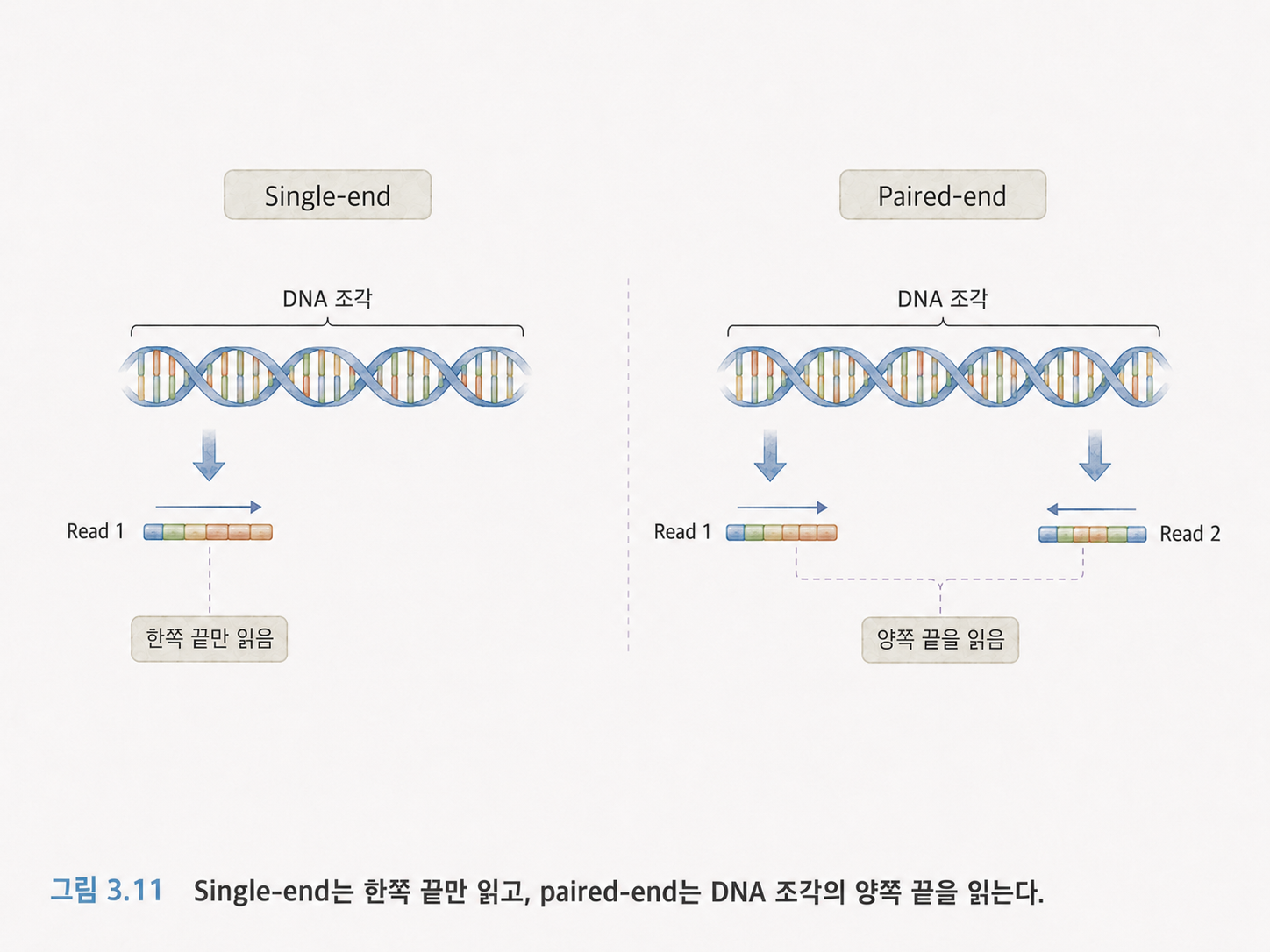
페어드 엔드는 정보가 더 많습니다. 양쪽 끝을 알면 리드가 표준 유전체의 어디에 붙는지 더 정확하게 판단할 수 있고, 삽입, 결실, 구조 변이를 찾는 데도 도움이 됩니다.
13. 정렬은 리드를 표준 유전체 지도에 붙이는 일입니다
시퀀싱 장비가 리드를 만들어도, 그 리드가 원래 유전체의 어느 위치에서 왔는지는 바로 알 수 없습니다. 그래서 표준 유전체에 리드를 맞춰봅니다. 이 과정을 정렬(Alignment)이라고 합니다.
예를 들어 리드가 다음과 같다고 해보겠습니다.
리드: ATGCAAGT
컴퓨터는 표준 유전체 전체에서 이와 가장 비슷한 위치를 찾습니다. 완벽히 일치하면 쉽지만, 실제로는 변이, 오류, 삽입, 결실이 있을 수 있습니다. 그래서 정렬 알고리즘은 약간의 불일치와 틈을 허용하면서 가장 그럴듯한 위치를 찾습니다.
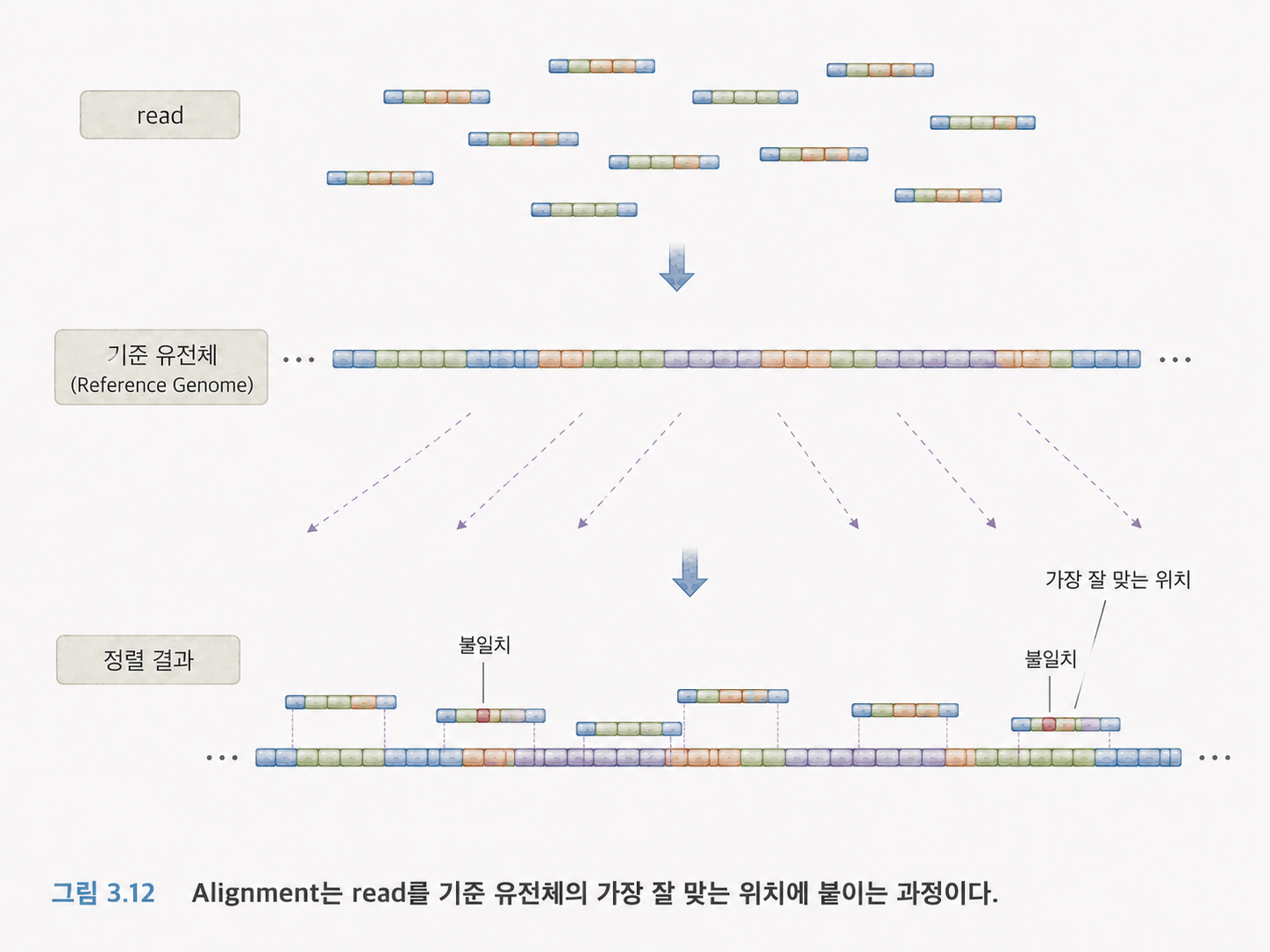
여기서 불일치(mismatch)는 글자가 다른 경우이고, 갭(gap)은 삽입이나 결실 때문에 중간에 빈칸을 넣어야 하는 경우입니다.
14. 문자열 비교의 기본은 거리와 점수입니다
정렬 알고리즘을 이해하려면 문자열 비교 감각이 필요합니다.
해밍 거리는 같은 길이의 문자열에서 서로 다른 위치가 몇 개인지 세는 방법입니다.
문자열 1: A T G C
문자열 2: A T T C
차이: G vs T → 1개
편집 거리는 한 문자열을 다른 문자열로 바꾸기 위해 필요한 최소 작업 수입니다. 여기서 작업은 삽입, 삭제, 대체입니다.
예를 들어 CAT을 CART로 바꾸려면 R 하나를 삽입하면 됩니다. 편집 거리 1이라고 볼 수 있습니다.
정렬 알고리즘은 단순히 같은지 다른지만 보지 않습니다. 일치하면 점수를 주고, 불일치나 갭에는 벌점을 주면서 가장 높은 점수를 얻는 정렬을 찾습니다.
15. 동적 프로그래밍은 큰 문제를 작은 표로 나눠 푸는 방법입니다
Needleman-Wunsch와 Smith-Waterman 알고리즘은 동적 프로그래밍을 사용합니다. 동적 프로그래밍이라는 말이 어렵지만, 기본 생각은 단순합니다.
큰 문제를 한 번에 풀기 어렵다면, 작은 문제들을 표에 차례로 기록하면서 최종 답을 찾는 방법입니다.
두 문자열을 비교할 때, 가로축과 세로축에 각각 문자열을 놓고 표를 만듭니다. 각 칸에는 “여기까지 비교했을 때 가장 좋은 점수”를 기록합니다. 마지막에는 표를 거슬러 올라가며 어떤 정렬이 가장 좋았는지 찾습니다.
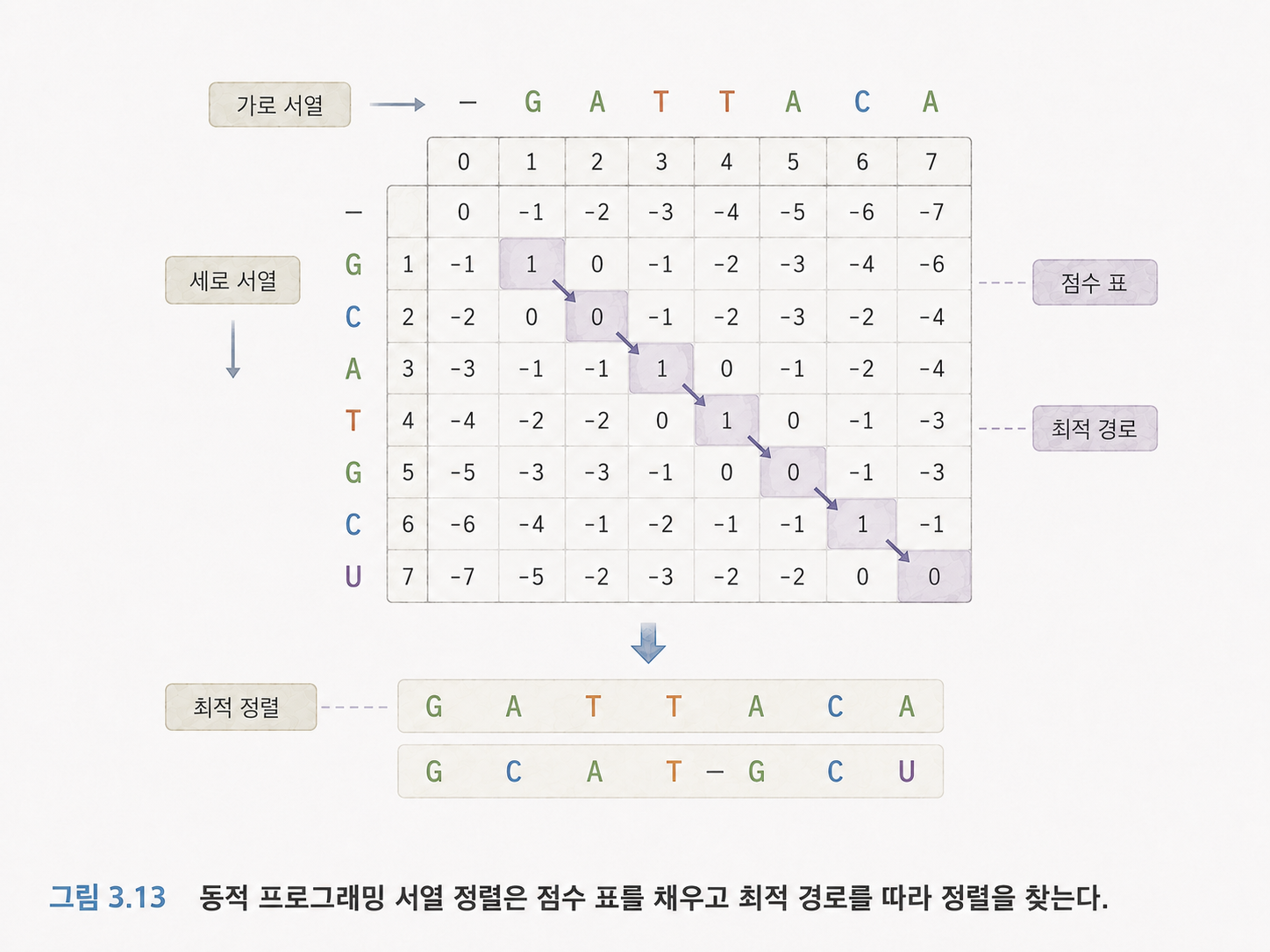
Needleman-Wunsch는 전역 정렬입니다. 두 문자열 전체를 처음부터 끝까지 맞추려는 방식입니다.
Smith-Waterman은 지역 정렬입니다. 두 문자열 중 가장 비슷한 일부 구간을 찾는 방식입니다.
NGS에서는 짧은 리드가 큰 유전체 어딘가에 붙어야 하므로, 지역적으로 비슷한 위치를 찾는 사고가 중요합니다.
16. BLAST는 빠르게 비슷한 서열을 찾는 검색 도구입니다
BLAST는 생물학 서열에서 비슷한 부분을 빠르게 찾는 대표적인 도구입니다. 모든 가능성을 완벽하게 하나하나 비교하면 너무 느리기 때문에, BLAST는 먼저 짧은 일치 조각을 찾고, 그 주변을 확장하는 식으로 빠르게 검색합니다.
여기서 휴리스틱이라는 말이 나옵니다. 휴리스틱은 완벽한 전수조사는 아니지만, 현실적인 시간 안에 꽤 좋은 답을 찾는 방법입니다.
BLAST 결과에서 E-value라는 값도 중요합니다. E-value는 이 정도로 좋은 결과가 우연히 나올 것으로 기대되는 횟수입니다. 값이 낮을수록 우연일 가능성이 낮고, 더 의미 있는 유사성으로 볼 수 있습니다.
17. k-mer는 DNA 문자열을 일정 길이 조각으로 자른 것입니다
k-mer는 길이가 k인 부분 문자열입니다. 예를 들어 ATGTAC이라는 서열에서 k=3이면 다음과 같은 3-mer가 나옵니다.
ATG
TGT
GTA
TAC
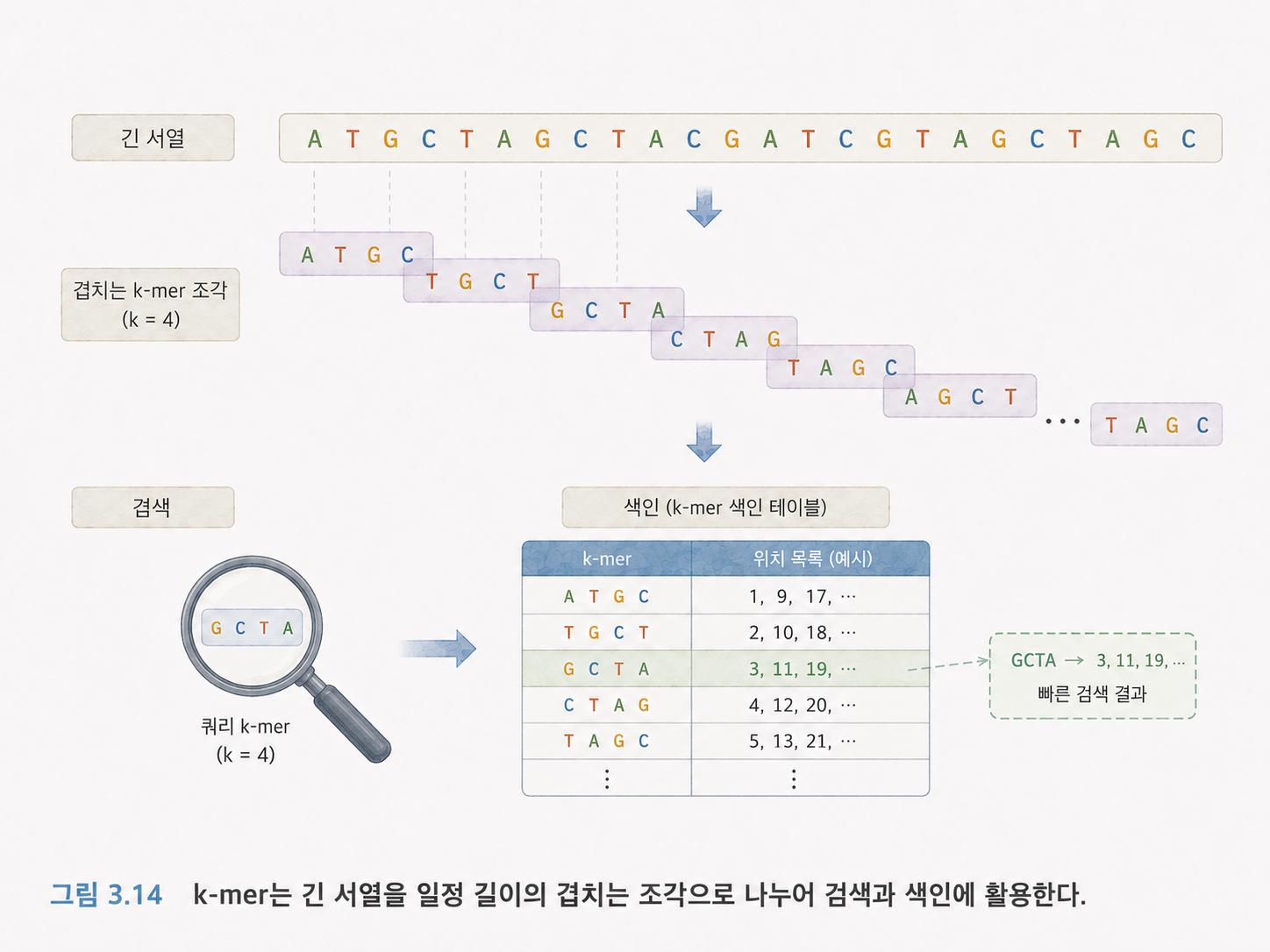
k-mer는 긴 DNA 문자열에서 빠르게 후보 위치를 찾는 데 쓰입니다. 모든 글자를 처음부터 끝까지 무식하게 비교하는 대신, 짧은 조각 단위로 먼저 검색하는 것입니다.
k가 크면 더 구체적인 조각을 찾을 수 있지만, 약간의 오류나 변이가 있을 때 놓칠 가능성이 커집니다. k가 작으면 더 많이 잡히지만, 너무 흔한 조각이 많아져 후보가 많아집니다.
18. 인덱스는 빠른 검색을 위한 책 뒤 색인입니다
3챕터에는 Burrows-Wheeler Transform, FM Index 같은 어려운 개념도 나옵니다. 이 부분은 처음부터 수학적으로 완벽히 이해하지 않아도 됩니다. 먼저 인덱스라는 감각을 잡으면 됩니다.
두꺼운 책에서 어떤 단어가 나온 위치를 찾는다고 해보겠습니다. 책 전체를 처음부터 끝까지 매번 읽으면 너무 오래 걸립니다. 대신 책 뒤에 색인이 있으면 단어가 나온 페이지를 빠르게 찾을 수 있습니다.
유전체 정렬에서도 비슷한 일이 필요합니다. 인간 유전체는 매우 길기 때문에 리드 하나하나를 전체 유전체와 직접 비교하면 비효율적입니다. 그래서 표준 유전체를 미리 검색하기 좋은 구조로 바꿔둡니다. BWT와 FM Index는 이런 빠른 검색을 가능하게 하는 방법입니다.
즉, 처음에는 이렇게 이해하면 됩니다.
BWT/FM Index는 거대한 유전체 문자열에서 리드가 붙을 후보 위치를 빠르게 찾기 위한 고급 색인 기술입니다.
19. SAM 파일은 정렬 결과를 담는 파일입니다
FASTQ가 “아직 지도에 붙이지 않은 리드”를 담는 파일이라면, SAM은 “표준 유전체에 붙인 뒤의 정렬 결과”를 담는 파일입니다.
SAM 파일에는 리드 이름, 어느 염색체에 붙었는지, 몇 번째 위치에 붙었는지, 정렬 품질은 어떤지, 실제 서열은 무엇인지 등이 들어갑니다.
중요한 필드 중 CIGAR 문자열이 있습니다. CIGAR는 리드가 표준 유전체와 어떻게 맞춰졌는지를 압축해서 표현합니다.
예를 들어 5M2I4M1D 같은 표현이 있을 수 있습니다.
- M은 match 또는 alignment match를 뜻합니다.
- I는 리드 쪽에 삽입이 있다는 뜻입니다.
- D는 참조 유전체 쪽에 비해 삭제가 있다는 뜻입니다.
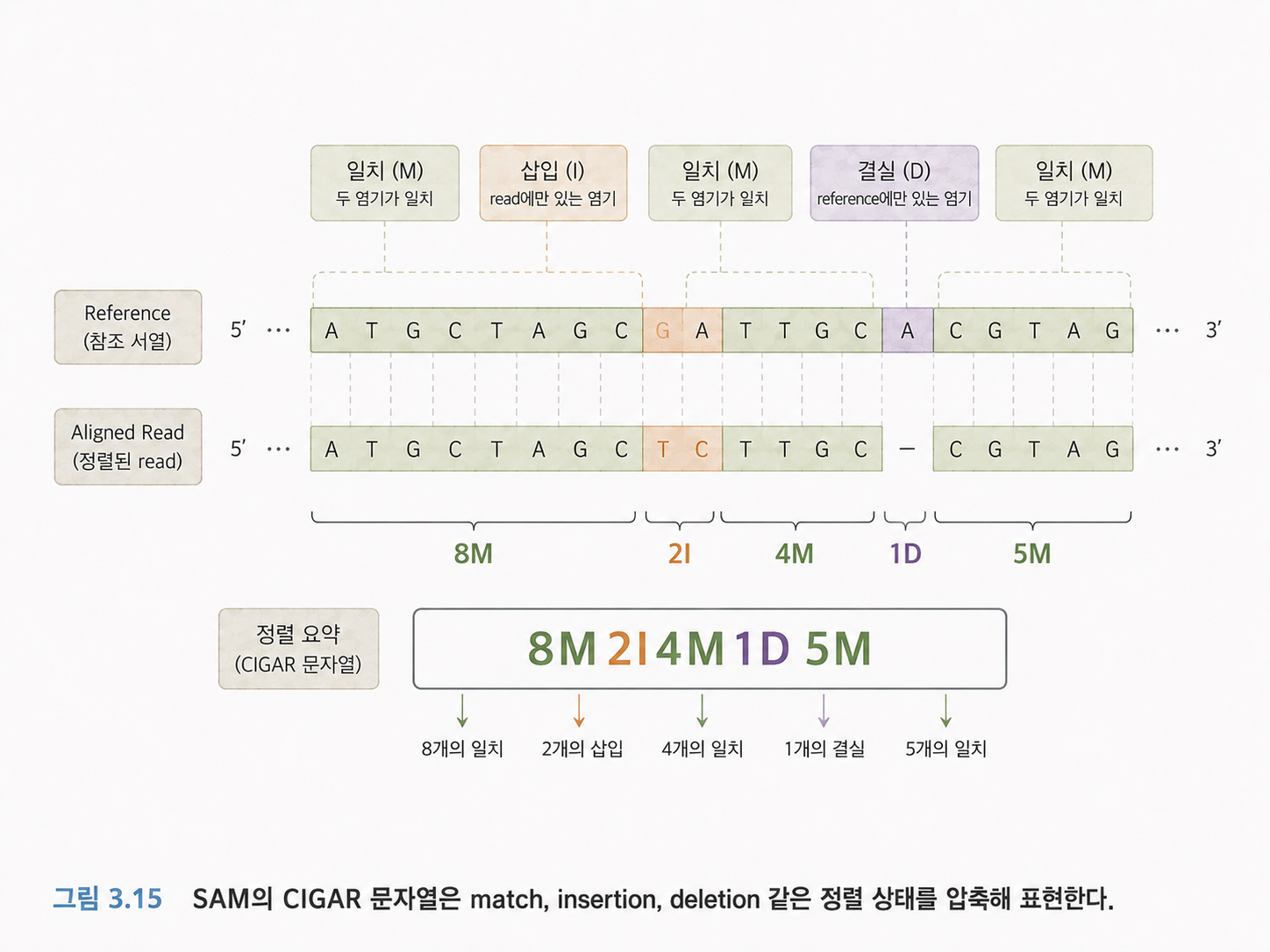
처음에는 CIGAR를 완벽히 해석하지 않아도 됩니다. 다만 “정렬이 단순히 위치만 말하는 것이 아니라, 어디가 맞고 어디가 삽입·결실인지까지 기록한다”고 이해하면 됩니다.
본편 진입 전 보강: Phred 점수는 오류 확률을 압축한 숫자입니다
3챕터 본편에는 Phred 점수 공식이 나옵니다.
Q = -10 × log10(P)
여기서 P는 염기를 잘못 읽었을 확률입니다. 로그 계산을 깊게 몰라도, 입문 단계에서는 아래 표를 먼저 익히면 충분합니다.

| Phred 점수 | 오류 확률 | 쉬운 해석 |
|---|---|---|
| Q10 | 1/10 = 10% | 10번 중 1번 정도 틀릴 수 있습니다. |
| Q20 | 1/100 = 1% | 100번 중 1번 정도 틀릴 수 있습니다. |
| Q30 | 1/1000 = 0.1% | 1000번 중 1번 정도 틀릴 수 있습니다. |
| Q40 | 1/10000 = 0.01% | 10000번 중 1번 정도 틀릴 수 있습니다. |
중요한 규칙은 이것입니다.
Q가 10 올라가면 오류 확률은 10분의 1로 줄어듭니다.
예를 들어 오류 확률이 0.01 = 10^-2라면 다음처럼 계산합니다.
Q = -10 × log10(0.01)
= -10 × log10(10^-2)
= -10 × (-2)
= 20
따라서 Q20은 오류 확률 1%에 해당합니다.
본편 진입 전 보강: k-mer 개수는 L - k + 1입니다
k-mer는 길이 k짜리 DNA 조각입니다. 길이 L인 문자열에서 k-mer를 한 칸씩 밀어가며 만들면 개수는 다음과 같습니다.
k-mer 개수 = L - k + 1
예를 들어 ATGTAC은 길이가 6입니다. k=3이면 3-mer는 다음 네 개입니다.
ATG, TGT, GTA, TAC
계산으로도 6 - 3 + 1 = 4입니다. 이 감각은 BLAST, 정렬 후보 탐색, 인덱싱을 이해할 때 필요합니다.
본편 진입 전 보강: 동적 프로그래밍 표는 “한 칸씩 점수를 고르는 표”입니다
Needleman-Wunsch나 Smith-Waterman 같은 정렬 알고리즘은 처음 보면 수학 표처럼 보입니다. 하지만 핵심은 각 칸에서 몇 가지 후보 점수 중 가장 좋은 값을 고르는 것입니다.
예를 들어 현재 칸의 문자가 서로 일치한다고 해보겠습니다.
왼쪽 위 값 = 2
왼쪽 값 = 0
위쪽 값 = 1
일치 점수 = +1
갭 벌점 = -2
현재 칸의 후보는 다음과 같습니다.
왼쪽 위에서 대각선 이동: 2 + 1 = 3
왼쪽에서 이동: 0 - 2 = -2
위쪽에서 이동: 1 - 2 = -1
가장 큰 값은 3이므로 현재 칸에는 3을 씁니다. 전체 알고리즘을 외우기보다, 본편의 행렬 예시를 볼 때 “각 칸은 이전 칸들에서 올 수 있는 후보 중 하나를 고르는구나”라고 이해하면 됩니다.
본편 진입 전 보강: CIGAR는 리드와 참조가 각각 얼마나 소비되는지 보여줍니다
CIGAR 문자열은 리드가 참조 유전체와 어떻게 맞는지를 압축해서 보여줍니다.
예를 들어 8M2I4M1D5M을 보겠습니다.
M 총합 = 8 + 4 + 5 = 17
I 총합 = 2
D 총합 = 1
- 리드에서 소비되는 염기 수: M + I = 17 + 2 = 19
- 참조 유전체에서 소비되는 염기 수: M + D = 17 + 1 = 18
삽입(I)은 리드 쪽 염기를 소비하지만 참조 위치는 소비하지 않습니다. 결실(D)은 참조 위치를 소비하지만 리드 쪽 염기는 소비하지 않습니다. 이 차이를 알아야 SAM/BAM 파일의 정렬 결과를 제대로 읽을 수 있습니다.
본편 진입 전 보강: BWT는 문자열을 정렬해 빠른 검색용 색인을 만드는 출발점입니다
BWT를 처음부터 완전히 증명할 필요는 없습니다. 다만 본편의 BANANA$ 예시를 따라가려면 “회전 → 정렬 → 마지막 열 읽기” 흐름을 알아야 합니다.

짧은 예로 ABA$를 보겠습니다. $는 문자열의 끝을 표시하는 특수 기호입니다.
모든 회전:
ABA$
BA$A
A$AB
$ABA
사전순 정렬:
$ABA
A$AB
ABA$
BA$A
마지막 열:
A, B, $, A
따라서 이 예시의 BWT 결과는 AB$A입니다. 실제 유전체에서는 이런 변환과 색인을 이용해 긴 참조 서열 안에서 리드가 붙을 후보 위치를 빠르게 찾습니다.
20. 3챕터 진입 전 핵심 정리
| 선수지식 | 아주 쉬운 설명 | 3챕터에서 필요한 이유 |
|---|---|---|
| 시퀀싱 | DNA/RNA 글자 순서를 읽는 기술입니다. | NGS 전체의 출발점입니다. |
| 리드 | 장비가 읽어낸 DNA 조각입니다. | FASTQ, 정렬, 조립의 기본 단위입니다. |
| Short Read | 짧은 조각을 많이 읽는 방식입니다. | Illumina 이해에 필요합니다. |
| Long Read | 긴 조각을 읽는 방식입니다. | Nanopore, PacBio, 유전체 조립 이해에 필요합니다. |
| 어댑터 | DNA 조각 양끝에 붙이는 손잡이입니다. | 라이브러리 제작과 플로우 셀 결합에 필요합니다. |
| 프라이머 | 읽기 또는 복사가 시작되는 출발점입니다. | Read 1, Read 2 이해에 필요합니다. |
| FASTQ | 리드 서열과 품질 점수를 담는 파일입니다. | 시퀀싱 데이터 처리의 기본 파일입니다. |
| Phred 점수 | 염기 호출의 신뢰도를 나타내는 점수입니다. | 품질 필터링과 트리밍에 필요합니다. |
| 정렬 | 리드를 표준 유전체 위치에 붙이는 일입니다. | 변이 찾기와 후속 분석의 출발점입니다. |
| k-mer | DNA 문자열을 길이 k 조각으로 나눈 것입니다. | 빠른 서열 검색과 정렬 후보 탐색에 필요합니다. |
| 인덱스 | 빠른 검색을 위한 색인 구조입니다. | 대용량 유전체 정렬을 빠르게 하는 데 필요합니다. |
| SAM/CIGAR | 정렬 결과와 정렬 방식을 기록하는 형식입니다. | 리드가 참조 유전체와 어떻게 맞는지 해석하게 해줍니다. |
| Phred 계산 | Q 점수가 10 올라가면 오류 확률은 10분의 1로 줄어듭니다. | 품질 점수와 오류 확률의 관계를 읽게 해줍니다. |
| 동적 프로그래밍 | 정렬 점수를 작은 표의 칸별 계산으로 채우는 방식입니다. | Needleman-Wunsch, Smith-Waterman 행렬 예시를 따라가게 해줍니다. |
| BWT/FM Index | 문자열을 빠른 검색이 가능한 색인 구조로 바꾸는 방법입니다. | 대용량 유전체 정렬이 왜 빠르게 가능한지 이해하게 해줍니다. |
문제 풀이
차세대 시퀀싱과 데이터 구조
주관식 답안은 Gemini API로 채점합니다. API 키는 이 브라우저에만 저장됩니다.
-
1. [쉬움] 객관식
시퀀싱의 기본 의미로 가장 적절한 것은 무엇인가?
-
2. [쉬움] 객관식
리드(read)에 대한 설명으로 가장 적절한 것은 무엇인가?
-
3. [쉬움] 객관식
Short Read와 Long Read를 구분하는 가장 기본 기준은 무엇인가?
-
4. [쉬움] 객관식
PCR의 기본 목적에 가장 가까운 것은 무엇인가?
-
5. [쉬움] 객관식
어댑터와 프라이머에 대한 설명으로 가장 적절한 것은 무엇인가?
-
6. [쉬움] 객관식
FASTQ 파일에 들어가는 핵심 정보로 가장 적절한 것은 무엇인가?
-
7. [쉬움] 객관식
Phred 점수의 의미로 가장 적절한 것은 무엇인가?
-
8. [보통] 객관식
Illumina 시퀀싱의 기본 원리로 가장 적절한 것은 무엇인가?
-
9. [보통] 객관식
Nanopore 시퀀싱의 설명으로 가장 적절한 것은 무엇인가?
-
10. [보통] 객관식
PacBio 방식의 특징으로 가장 적절한 것은 무엇인가?
-
11. [보통] 객관식
정렬(alignment)에 대한 설명으로 가장 적절한 것은 무엇인가?
-
12. [보통] 객관식
k-mer에 대한 설명으로 가장 적절한 것은 무엇인가?
-
13. [보통] 객관식
인덱스가 대용량 유전체 분석에서 중요한 이유는 무엇인가?
-
14. [어려움] 객관식
다음 FASTQ 조각에서
+줄 바로 다음 줄의 의미로 가장 적절한 것은 무엇인가?@read_001 ACGTACGT + IIIIHHHH -
15. [어려움] 객관식
Phred 점수 Q가 높다는 말의 해석으로 가장 적절한 것은 무엇인가?
-
16. [어려움] 객관식
유전체 조립과 참조 유전체 정렬의 차이에 대한 설명으로 가장 적절한 것은 무엇인가?
-
17. [어려움] 객관식
BLAST가 “빠른 검색 도구”로 이해되는 이유로 가장 적절한 것은 무엇인가?
-
18. [어려움] 객관식
다음 CIGAR 문자열
8M2I4M1D5M에 대한 해석으로 가장 적절한 것은 무엇인가? -
19. [어려움] 객관식
페어드 엔드 시퀀싱의 장점으로 가장 적절한 것은 무엇인가?
-
20. [어려움] 객관식
다음 중 3챕터의 핵심 흐름을 가장 적절히 나타낸 것은 무엇인가?
-
21. [보통] 객관식
계산형: Phred Q30에 해당하는 오류 확률로 가장 적절한 것은 무엇인가?
-
22. [보통] 객관식
계산형: 오류 확률 P=0.01일 때 Q=-10×log10(P)이다. Phred 점수는 얼마인가?
-
23. [보통] 객관식
계산형: Phred 점수가 Q20에서 Q30으로 10 올라가면 오류 확률은 어떻게 변하는가?
-
24. [보통] 객관식
계산형: 서열
ATGTAC의 길이는 6이다. k=3일 때 가능한 3-mer 개수는? -
25. [보통] 객관식
계산형: 길이 10인 DNA 서열에서 k=4인 k-mer는 몇 개인가?
-
26. [보통] 객관식
계산형: CIGAR
8M2I4M1D5M에서 M의 총합은 얼마인가? -
27. [보통] 객관식
계산형: CIGAR
8M2I4M1D5M에서 리드가 소비하는 염기 수는 얼마인가? -
28. [보통] 객관식
계산형: CIGAR
8M2I4M1D5M에서 참조 유전체가 소비되는 길이는 얼마인가? -
29. [보통] 객관식
계산형: 정렬 DP에서 왼쪽 위 2, 왼쪽 0, 위쪽 1이고, 문자가 일치해 +1, 갭 벌점 -2라면 현재 칸의 최댓값은?
-
30. [보통] 객관식
계산형:
ABA$의 회전을 정렬했을 때 마지막 열이 A, B, $, A라면 BWT 결과는? -
31. [쉬움] 객관식
FASTQ 한 리드 기록의 기본 4줄 구성으로 가장 적절한 것은 무엇인가?
-
32. [보통] 객관식
BLAST의 E-value가 낮다는 말의 의미로 가장 적절한 것은 무엇인가?
-
33. [보통] 객관식
페어드 엔드 시퀀싱의 장점으로 가장 적절한 것은 무엇인가?
-
34. [보통] 객관식
계산형: Phred Q40의 오류 확률로 가장 적절한 것은 무엇인가?
-
1. [쉬움] 주관식 · Gemini 채점
리드와 FASTQ 파일의 관계를 설명하라.
-
2. [보통] 주관식 · Gemini 채점
Short Read와 Long Read의 장단점을 비교하라.
-
3. [보통] 주관식 · Gemini 채점
품질 점수와 어댑터 트리밍이 왜 필요한지 설명하라.
-
4. [어려움] 주관식 · Gemini 채점
k-mer와 인덱스가 빠른 서열 검색에 도움이 되는 이유를 설명하라.
-
5. [어려움] 주관식 · Gemini 채점
정렬 결과를 해석할 때 SAM/CIGAR 같은 형식이 필요한 이유를 설명하라.